Understanding Permanent Marks and Methods for UDI Marking and Verification

Contributed by | Microscan
Direct Marking for FDA UDI Compliance
Direct marking is an important component of the FDA Unique Device Identification (UDI) regulation. The purpose of UDI is to establish a standard method for identifying and tracing medical devices throughout their lifecycles – from production, to distribution, to use. For medical devices that are reused many times during their lifecycles, a UDI must be able to withstand any amount of handling or reprocessing. The most reliable way to ensure a UDI lasts the lifecycle of a multi-use device is by affixing a permanent mark to the device itself, rather than a temporary package or label. Direct part marking (DPM) is not a new concept in industrial manufacturing, but is relatively new territory for medical device identification. For manufacturers now under obligation to comply with FDA UDI requirements by directly marking devices, this white paper offers current FDA guidance as well as answers to the most common questions, including:
⦁ What is Unique Device Identification?
⦁ What does it mean to “permanently mark” a device?
⦁ Which medical devices require direct marks?
⦁ What are the deadlines for permanent UDI marking?
⦁ Does the FDA require a specific marking method?
⦁ What can be done to ensure mark quality and compliance?
What is Unique Device Identification?
On September 24, 2013, the U.S. Food and Drug Administration (FDA) established a unique device identification system to identify and trace medical devices throughout their distribution and use. UDI is expected to improve patient safety and healthcare industry processes greatly by implementing a global system of standards for device identification. This system makes it possible to identify devices and attributes of devices in the market quickly and accurately, ensuring safety and effectiveness. In the case of adverse events such as a product recalls, devices can be rapidly and precisely identified to take corrective action, minimizing risk to patients and consumers.
As part of the FDA UDI mandate, medical devices must bear a unique device identifier (UDI) code in human-readable (text) and machine-readable (barcode, RFID) format on labels or packaging (the UDI code may be provided in either or both human- or machine-readable format for direct marks). A UDI is a unique numeric or alphanumeric code that consists of:
⦁ DI (Device Identifier): A mandatory fixed portion that identifies the device and device labeler.
⦁ PIs (Production Identifiers): Conditional, variable portions that denote data like batch numbers and expiration dates.
.jpg)
A fictitious example of a medical device label that conforms to the requirements of the FDA’s UDI (Unique Device Identifier) initiative for device labels and packaging.
For the purpose of maintaining standardization across all devices bearing UDI, a manufacturer’s DI must be issued under a system operated by an FDA-accredited issuing agency such as:
- GS1: www.gs1.org
- Health Industry Business Communications Council (HIBCC): www.hibcc.org
- International Council for Commonality in Blood Banking Automation (ICCBBA): www.iccbba.org
Labelers of medical devices must submit the DI from each device’s UDI code to the FDA’s Global Unique Device Identification Database (GUDID) to log the device for global traceability.
There are three fundamental elements to ensuring the effectiveness of UDI implementation:
- A globally-standardized UDI code structure;
- A single, global database of all existing UDI codes;
- The ability to identify a device at any point in its lifecycle using the standardized UDI codes as cross-referenced with the global database.
A UDI must be properly created, submitted, and affixed to a device to ensure absolute compliance with the FDA UDI regulation. While the creation and submission of UDI codes follow a defined set of steps outlined above, the process of affixing UDI codes greatly depends on the device and its intended use. A device must bear the same UDI code throughout its lifecycle (from manufacture to disposal), regardless of handling, reprocessing, or reuse.
What Does It Mean to “Permanently Mark” a Device?

A pacemaker has been permanently marked with a Data Matrix symbol using a method of direct part marking (DPM) called laser etch.
As a requirement of the FDA UDI regulation under 21 CFR 801.45 (the Code of Federal Regulations for medical device marking): “[A] device that must bear a unique device identifier (UDI) on its label must also bear a permanent marking providing the UDI on the device itself if the device is intended to be used more than once and intended to be reprocessed before each use.” This requirement applies to medical devices of every FDA classification (Class I, II, or III) that are intended to be used over long periods of time. Since reprocessing devices may cause devices to become separated from their original labels and packages, direct marking is necessary to ensure that a UDI is permanently available through a device’s distribution and use, even where packaging and labels are unavailable.
Permanent marks are also known as direct part marks (DPM), and are used widely in industrial part tracking from electronics manufacturing to automotive assembly. These marks are affixed directly to parts by abrading a part surface or marking in some other manner that cannot be discarded, torn, obscured, wiped off, or easily degraded. An example of an impermanent mark (not permanent) is an inkjet code on a paper label or package, which can be removed from the device, damaged by physical contact, or distorted by moisture, temperature, and other elements. An example of a permanent mark is a code that is etched directly onto the surface of a device, by a laser for example, which removes the surface layer of the substrate of a device to expose the code in a varying color or contrast. Other types of permanent marking include electrochemical etch and dot peen. These methods can also be used to abrade codes into identifier tags or plaques that can then be permanently affixed to a device.
.jpg)
An example of a laser-etched direct part mark (DPM) code on a PCB. The laser has revealed a contrasting color of the PCB’s substrate, allowing the code to be distinguished from the surface of the part.
Direct part marks are considered “permanent” because they are intended to last as long as the device itself, providing a means of device identification through the entire device lifecycle from manufacture to distribution to use and reuse. This is known as “cradle-to-grave traceability” and it ensures long-term product location in the supply chain (where was the part’s code scanned last?), product authenticity verification (who is the manufacturer indicated in the code data?), and responsiveness to adverse events like product recalls (which set of products were manufactured during the time that an adverse event took place, and where are the products now?).
.jpg)
Reading codes on products throughout manufacture, distribution, and use enables anyone to obtain “cradle-to-grave traceability” about where a product has been, where it is now, and where it is going.
It would require significant force to damage, remove, or otherwise render a direct mark illegible. For medical devices that must undergo reprocessing for multiple uses, direct part marks are required to ensure that they do not lose their identifying information throughout device lifecycles. Packaging and labels bearing UDI may be separated from devices over time, but direct part marking ensures that a device will always be identifiable through the global UDI database (GUDID).
What Is the Current FDA Guidance for UDI Direct Marking?
In June 2015, the FDA issued draft guidance for labelers of medical devices who are preparing to meet deadlines for marking devices with permanent UDI marks as part of the FDA UDI requirements.
Unique Device Identification: Direct Marking of Devices Draft Guidance: www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM452262.pdf
This document provides manufacturers with answers to preliminary questions, and can be used by device labelers to prepare for the final parameters of permanent marking and quality assurance when they are established. Once finalized, this document will provide a foundation for FDA and issuing agency regulations regarding permanent UDI marks on medical devices, their marking methods, tolerances for mark quality, and how to verify mark quality against issuing agency specifications to ensure UDI compliance. Manufacturers can use an alternative approach to permanent marking if it satisfies the requirements of the currently-documented regulations. To ensure that an alternate approach satisfies UDI requirements, a manufacturer should contact the FDA staff responsible for this guidance listed on the document title page.
In its draft guidance, the FDA does not provide a specific approach to permanent medical device marking, citing the wide variety of existing devices, use conditions, and reprocessing methods that may require special considerations. The FDA stipulates only that a permanent UDI mark must contain all required information (meaning DI and PI data, as with impermanent UDI labels and packaging), and that the mark should be able to endure the expected use of a device throughout its lifecycle, including reprocessing. Currently, labelers can decide the most appropriate direct marking method for their device based on substrate, device type, intended use, and reprocessing method.
Which Medical Devices Require Direct Marks?
Under 21 CFR 801.45, a device that is required to have a UDI label (impermanent mark) must also be permanently marked with the UDI code if:
⦁ The device is intended to be used more than once.
⦁ The device is intended to be reprocessed before each use.
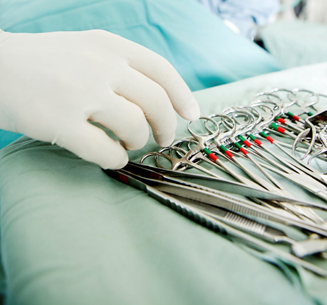
Surgical instruments that are used several times and reprocessed between each use are required to bear a permanent mark to comply with the FDA UDI regulation.
Unless the FDA has granted an exception to the manufacturer, the full UDI code (complete with agency-issued DI and all manufacturer-defined PI portions) must be directly marked on the device itself.
Standard FDA-approved exceptions are as follows:
⦁ Production identifiers (PI) are not required in the encoded data of UDI codes for Class I devices.
⦁ Class I devices that bear a UPC code on the device label and packaging are not required to comply with UDI direct marking requirements at all.
Use the FDA’s Product Classification Database to look up the class of a particular device:
What Is “Reprocessing”?
Reprocessing is defined as any process used to render a medical device, which has previously been used or contaminated, fit for subsequent reuse. Reprocessing is generally intended to remove blood, tissue, biological debris, and other contaminants, and to inactivate infectious microbes with the purposes of making devices safe for use on the next patient. According to FDA UDI regulations, devices that require reprocessing are generally intended for repeated uses on or by more than a single patient. For example, surgical devices.

After a sterilization process, a tray of surgical instruments may be used for a new patient.
If a device is intended to be used only once before disposal, or used multiple times by the same patient (not different patients), the device does not need to be directly marked with a UDI code.
What Are the Deadlines for Permanent UDI Marking?
The FDA outlines the following deadlines for directly marking medical devices with UDI codes, which are separate deadlines than those established for UDI labels and packaging.
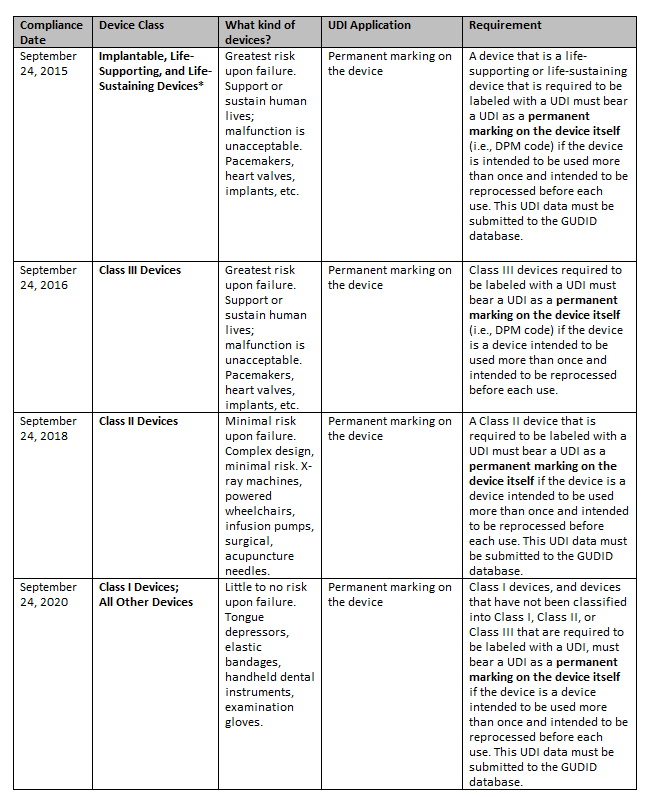
* The FDA recommends that labelers search the CDRH Product Classification database for the most current information on FDA product codes to determine if devices are considered implantable, life-sustaining, or life-supporting.
Full compliance dates and requirements can be viewed at www.fda.gov.
For devices classified through the De Novo Classification Process or cleared in a 510(k) submission, the FDA states that manufacturers must conduct analysis or testing to identify whether the process of directly marking a device would affect the safety and effectiveness of a device. If a safety issue is determined to exist, the manufacturer can then apply for an exception to UDI direct marking requirements, or proceed to mark the device provided that the manufacturer receives clearance of a new 510(k) submission.
What Part of the UDI Must Be Marked on a Device?
As with device labels and packaging, a UDI must contain all information required by the FDA (agency-issued DI and all required manufacturer-defined PIs) in human- or machine-readable format. A directly-marked UDI must be identical to the UDI code that appears on the device label or packaging, unless a manufacturer chooses to distinguish the packaged device from the unpackaged device. In addition to the DI issued for its labels and packaging, a manufacturer may request to be issued a second DI for its directly-marked UDI code, where the DI issued for the label and packaging is known as the primary DI, and the DI issued for the direct mark is known as the direct-mark DI (DM-DI).
For any medical device, each unique DI issued for a device must be submitted to the GUDID for compliance with FDA UDI documentation. If the primary DI and the DM-DI for a device are the same, only the primary DI needs to be submitted. If the primary DI and DM-DI are different, then the manufacturer must also submit the DM-DI to the GUDID. Upon submission of their DIs to the GUDID, the manufacturer must indicate whether the primary DI and DM-DI are the same or different for a given device.
What Is the Required Format of a UDI Mark?
Unlike UDI codes on labels and packaging, when a UDI is directly-marked on a device, the UDI code may be provided in either or both of the following formats:
⦁ Human-readable: Easily-legible, plain-text format.
⦁ Machine-readable: Able to be interpreted by automatic identification and data capture (AIDC) technology, such as barcode readers, machine vision systems, RFID equipment, or any other technology that will provide the UDI code to databases on demand.
These format options for UDI direct marks are provided to accommodate devices that have limited surface area or other constraints regarding how and where marks can be applied.
.jpg)
On small implantable devices, surface area may be limited. High-density symbols can be marked on devices to contain long strings of data using the smallest possible footprint.
Formatting requirements vary by the issuing agency (GS1, HIBCC, ICCBBA) and must be strictly adhered to in order to meet compliance for the agency’s specifications. It is important to understand the specifications of each issuing agency and establish a testing process to ensure that UDI marks continue to comply. Testing can be accomplished using technology like a barcode verifier or other standards-based verification method. For reliability and ease of device identification, a machine-readable format should be used to ensure the highest-precision UDI decoding at a reduced margin of error (such as human error in interpreting human-readable marks), as well as standards-based verification by specially-engineered software for ensuring compliance.
Which Data Carriers Should Be Used for Machine-Readable Marks?
Machine-readable UDI codes are encoded into what are called “data carriers” (the machine-readable media that carries the UDI data; for example, barcodes, RFID chips, or other media). The FDA does not specify which data carrier must be used by the manufacturer for UDI compliance, but considerations should be made regarding issuing agency specifications for a given manufacturer or device, the types of data carriers the manufacturer’s customers are able to read, and the types of data carriers that can be easily marked and read on medical devices.
2D symbols such as Data Matrix are the most common data carriers used for direct part marking because of their small size, high data capacity, and built-in error correction. Data Matrix symbols consist of an arrangement of small dots or squares used to encode data. Compact 2D symbols like these are more easily and accurately marked on devices than lines of text or one-dimensional codes like linear barcodes. 2D symbols can also be marked by a variety of methods (laser, dot peen, or electrochemical etch).
.jpg)
The data-carrying capacity of Data Matrix symbols makes it easy to accommodate large data volumes in small spaces, like the surface areas of some medical devices.
Data Matrix symbols are extremely reliable and can be read in any orientation, at low or high contrast, and even when errors are present. The symbology standard for Data Matrix includes error-detection and correction algorithms to ensure reliable reading despite parts of the symbol being obscured or missing. This is a helpful feature in environments where symbols may be obscured by grease, dirt, paint and chemical coatings, and when the symbology is applied to metal and other reflective surfaces. As a result of error-correction, Data Matrix symbols with damage, distortion, or minor defects can still be decoded accurately, even if more than twenty percent of the symbol is obscured.

Built-in error correction allows Data Matrix symbols to be decoded despite damage or partial obstruction for the most reliable part identification throughout processes.
Does the FDA Require a Specific Marking Method?
For the purposes of complying with the FDA UDI regulation, direct marking is defined simply as the process of affixing a UDI permanently to a device that requires a UDI code. There is no specified method for affixing a permanent UDI to a device, except that the FDA requires that:
⦁ The permanent UDI comply with the requirements of 21 CFR 801.45.
⦁ The permanent UDI must last throughout the device lifecycle, including usage and reprocessing.
According to the FDA, “possible methods to directly mark a device with a UDI include etching [laser, dot peen, or electrochemical etch], attaching a permanent plaque to durable equipment, or affixing a permanent tag such as a radio frequency identification (RFID) tag to the device.” The precise method is left to the manufacturer given the variety of devices in the market, as well as possible reprocessing methods that devices may undergo during their lifecycles. This also leaves room for new marking methods, device usages, or reprocessing methods that result from technology developments in the future. In general, whatever marking method the manufacturer chooses, the device must bear a permanent mark that is legible by either humans or machines throughout its lifecycle.

A dot peen direct part marking system uses a metal stylus to indent the elements of a 2D symbol onto a metal surface.
If the UDI on a Label Changes, Does the Directly-Marked UDI Need to be Replaced?
Once a device has been marked with a UDI code and the code has been verified to be in compliance with UDI direct marking requirements, there is no FDA mandate to replace the UDI direct part mark even if the UDI that appears on the device label or packaging changes.
Do Company Names or Part Numbers Marked Directly to Devices Satisfy the UDI Requirements?
Only the UDI itself, in human- or machine-readable format, can satisfy UDI direct marking requirements. The company name, part number, catalog number, or other data cannot be used in place of an issued and submitted UDI code. Manufacturers who choose to mark other information on their devices should take care to leave enough space to also mark the UDI. Lack of space for a UDI mark due to other markings on the device will not be sufficient justification for exemption from the UDI regulation for direct marking.
Can a Labeler Voluntarily Comply with Direct Marking Requirements?
Although not required for every device, the FDA encourages manufacturers to mark all medical devices in permanent format to ensure identifying information is widely available throughout device lifecycles. If a labeler decides to mark a UDI directly to a device without an FDA requirement or prior to the UDI marking deadline for the device, submission of the device’s issued DI to the GUDID would also be voluntary.
Are there Exceptions to the UDI Direct Marking Rule?
There are four cases in which the UDI direct marking requirement does not apply:
⦁ If the type of mark interferes with the safety or effectiveness of the device.
⦁ If the device cannot be directly marked because it is not technologically feasible.
⦁ If the device is a single-use device (SUD) and is subjected to additional processing and manufacturing for the purpose of an additional use.
⦁ If the device has been previously marked under 21 CFR 801.45 (a).
A “single-use device” (SUD) is defined as a device that is intended for one use, or for multiple uses on a single patient during a single procedure. A device intended for single-use that is subjected to additional processing and manufacturing for the purpose of an additional single use on another patient should not require direct marking. However, such a reuse of a single-use device would generally require additional clearance or approval from the FDA unless exempt, as well as compliance with general UDI labeling and data submission requirements by the entity performing the additional processing and manufacturing for the purpose of an additional use of the device.
More information on data submission requirements (510(k)) for reprocessed single-use devices can be found on the FDA website:
Understanding Needs for Specific Applications
Each application of UDI, depending on the specific device and device use, may require a unique method of marking, or a unique string of data to be encoded into the UDI. It is important to understand the requirements of the UDI issuing agency, including which data should be encoded, how it should be formatted, and the parameters to which a directly-marked UDI must adhere. A UDI mark is permanently affixed, so precautions should be taken to ensure that a mark, such as a laser etch or other abrasion, is not marked improperly or with incorrectly-encoded data. Poor marks can result in great expense to the manufacturer or labeler, including materials loss, process downtime, noncompliance fees, or legal action.
Challenges with Reading Direct Marks
Whether printed or directly-marked, codes that are low-contrast or marked on highly-reflective surfaces can be challenging to decode. Directly-marked codes pose special challenges because they are applied in a variety of ways to surfaces of varying reflectiveness, contrast, color, texture, and curvature. These conditions result in reflections and shadows that can obstruct areas of the code or yield poor contrast, making it difficult for barcode readers to distinguish codes against device surfaces. Without proper lighting to ensure code contrast, even the best barcode readers may have trouble decoding a high-quality mark. This challenge is further complicated by the fact that manufacturers cannot control the equipment their customers will use to read the codes on their devices.

A directly-marked dot peen Data Matrix symbol on this curved metal part exhibits low contrast (blending in with the surface material) when viewed under ambient light.
It is especially important, therefore, that marks are produced at the highest level of quality possible (maximum contrast, consistent size, and consistent depth of light and dark elements) to ensure reliable decoding by the widest range of barcode readers and cameras used from manufacturer to consumer. A UDI code that cannot be read at any given point in its lifecycle cannot be identified and cross-referenced with the GUDID, leaving room for substantial risk should an adverse event require that action be taken for a device or series of devices. For example, if an implantable device with a direct UDI mark were unable to be identified before implantation, and then later recalled, a healthcare provider wouldn’t know to contact patients who received the device with this potentially life-saving information.
What Can Be Done to Ensure Mark Quality and Compliance?
A handheld verification system is used to check both the quality of a UDI code and the accuracy of the encoded data structure according to issuing agency specifications.
Verification systems are specially-engineered equipment that use imaging technology, calibrated lighting, and software to perform precise data validation processes and visual measurements of marks to guarantee quality and compliance. Verification systems can be used before or during the application of a UDI code to validate that data within a UDI code is properly formatted according to issuing agency specifications, and that the UDI code will be legible throughout distribution and use. It is important to implement a verification step in UDI operations to avoid the risk of noncompliance and to ensure auditable processes.
Advanced verification systems offer built-in software with simple graphical interfaces programmed with UDI issuing agency specifications. Manufacturers simply select their application standard in the software and the verification system automatically recognizes their code type (data carrier) and applies the necessary legibility grading and data formatting specifications for the code based on issuing agency requirements from GS1 to HIBCC. For example, these software programs can automatically separate the segments of a UDI code into GS1 application identifiers (AIs) and ensure proper data structure according to GS1. These systems verify that AIs are valid, and that the encoded data matches the prescribed format (for instance, 14 characters required for a GTIN). Some verification systems offer enhanced or add-on features that allow manufacturers to program specific data strings into the software, ensuring that decoded strings from their UDI codes match the intended set of characters (what is known as a match string process).

A handheld verification system is used to check both the quality of a UDI code and the accuracy of the encoded data structure according to issuing agency specifications.
In addition to checking encoded data structure, verification systems also assign grades (A-F) to codes according to how well they meet industry standards for barcode print or mark quality to ensure long-term legibility by AIDC equipment. ISO/IEC barcode quality standards (ISO/IEC 15416 and ISO/IEC 15415) specify methodologies for grading printed 1D barcodes and 2D symbols on labels and packaging. It is best practice to verify that printed codes meet a grade of C or higher to ensure compliance with industry standards and long-term readability throughout the supply chain.
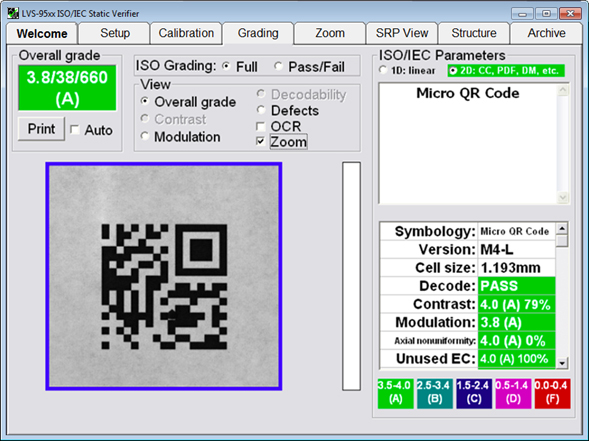
An image of verification software with built-in ISO/IEC parameters for grading barcode print quality compliance to standards. The above Micro QR Code has achieved an “A” grade.
Due to the unique conditions of direct part marks, a third specification – ISO/IEC TR 29158 – is used to assess the quality of direct part marks (DPM). This technical recommendation not only employs the basic ISO/IEC barcode quality parameters used for printed codes, but also specifies how lighting must be used to illuminate a marked code properly for verification, accounting for reflections and shadows due to various marking methods, materials, and surface characteristics. The ISO/IEC TR 29158 barcode quality specification was created to act as a bridge between existing barcode quality specifications and unique DPM environments in order to provide a standardized, image-based measurement that can closely predict the ability of a barcode reader to decode a symbol reliably based on a mark’s quality. As with printed codes, directly-marked codes should receive at least a C grade to guarantee their legibility throughout device lifecycles.
Now that the FDA has mandated the implementation of UDI direct marks, UDI verification methodologies will begin to incorporate verification parameters like those specified by ISO/IEC TR 29158 to ensure that each UDI meets issuing agency requirements for directly-marked codes. Although these agency requirements are still being finalized for direct part marks, manufacturers should prepare to apply such methodology to their operations to certify the validity and quality of their UDI codes in both print and mark format. Medical device manufacturers should choose a verification system that offers ISO/IEC quality standards for both printed and marked codes to account for the full range of UDI implementation methods. These systems should also be capable of validating that the encoded data of printed UDI codes match that of their marked UDI codes. For example, ensuring that the application identifiers (AIs) encoded within a GS1-formatted UDI code are the same in both printed and marked format. Manufacturers should be careful to select a verification system engineered for DPM codes, as these codes have unique reading and verification requirements.
Why Is Choosing a DPM Verifier so Important?
The key to both reading and verifying readability of direct UDI marks is lighting. When a code is illuminated, light reflects differently based on the substrate that it hits and the abrasions on the substrate surface. Verification systems distinguish and measure the variances between light and dark elements of a code (bars and spaces in a linear barcode, and dark cells and light cells in a 2D symbol). A directly-marked code poses unique challenges because the reflectance of light and dark elements in a code depends on the uniformity of lighting to reveal a mark contrasted against its substrate. Generally, surfaces must be oriented toward lighting (or vice versa) such that the angle of incidence (the angle of light cast on the part) is equal to the angle of reflection (the angle of light returning to the barcode reader or verifier). For DPM codes, light reflections vary based on the marking method and the angle of lighting applied. Therefore it is difficult to obtain a “controlled” environment in which to verify the quality of a mark across all marking methods and substrates. Because of these unique lighting needs, code quality standards organizations such as ISO/IEC prescribe precise lighting angles and integration geometries for DPM verifiers to meet the greatest range of conditions. As UDI issuing agencies continue to refine their specifications for DPM code quality, DPM verification systems continue to emerge with more specific lighting geometries for standards-based grading of directly-marked codes, including UDI.
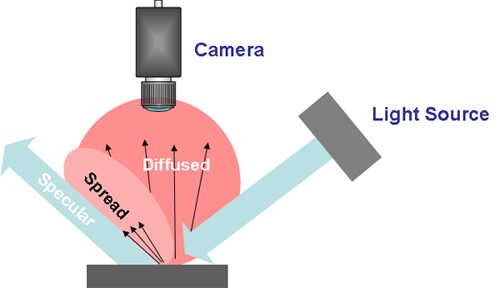
A camera “sees” a direct part mark in various contrasts depending on how light is reflected from the mark. This is affected by the angle of incidence (light hitting a mark), as well the features or inconsistencies of the mark or the device surface affecting the angle of reflection.
Best Practices for Ensuring UDI Quality and Compliance
Regardless of the print or marking method, when creating a code for UDI-regulated medical devices, it is important to always check the accuracy of encoded data before applying codes to final packaging or devices. First, print the code on any desktop or label printer and validate the accuracy of the encoded data structure using a verification system. Once the encoded data and structure is verified to meet the issuing agency specifications for UDI compliance, the code can then be etched or permanently affixed to a device using the manufacturer’s preferred marking method. Verifying code data prior to permanent marking can prevent costly quality issues later in the supply chain, such as customer fees, noncompliance issues, and scrap or rework, due to devices being marked with improperly-structured UDI codes.
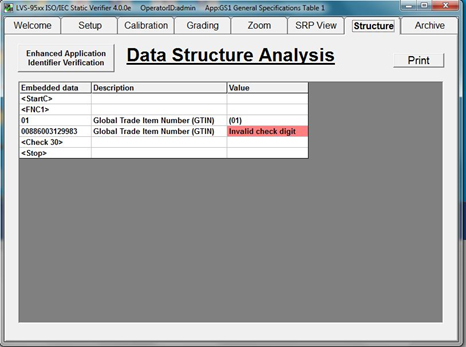
Verification Software automatically parses UDI codes to confirm whether encoded data is accurate and meets issuing agency specifications for the device in question. If an error in data structure is found, the verifier highlights the error and manufacturers can take steps to correct the code contents before further devices are marked with incorrect data.
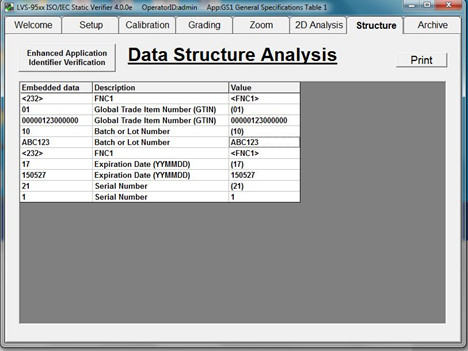
Once encoded UDI data strings are verified to contain correct information in the correct format, codes can be applied by permanent marking methods to medical devices on a broad scale.
Conclusion
Uniquely identifying devices with permanent marks is a new process for many medical device manufacturers, and the pressure to meet short deadlines for permanent device marking in compliance with the FDA UDI regulation has quickly become the device manufacturer’s greatest challenge. As agencies and solution providers alike work to define best practices for ensuring the readability of device codes in some of the most difficult reading conditions, manufacturers can best prepare for their compliance deadlines by arming themselves with knowledge from current documentation about UDI requirements and DPM solutions, and taking simple steps toward enhancing operations with marking methods. For manufacturers who have already implemented UDI on labels and packaging, the path forward to DPM implementation is now just a matter of selecting a mark type (data carrier), marking method, and process for verifying code accuracy and legibility (verification system). If a manufacturer chooses a marking method that best suits their device, a data carrier that best accommodates their marking method, and a verification system that is geared for standards-based grading based on UDI issuing agency requirements, the manufacturer’s operations will be well-prepared to meet FDA UDI direct marking requirements when they are finalized.
References
⦁ FDA. (2015, April 1). CFR – Code of Federal Regulations Title 21. Retrieved April 29, 2016 from https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=801.45
⦁ FDA. (2006, September 25). Guidance for Industry and FDA Staff - Medical Device User Fee and Modernization Act of 2002, Validation Data in Premarket Notification Submissions (510(k)s) for Reprocessed Single-Use Medical Devices. Retrieved April 29, 2016 from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm071434.htm
⦁ FDA. (2016, April 25). Product Classification. Retrieved April 29, 2016 from https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
⦁ FDA. (2015, June 26). Unique Device Identification: Direct Marking of Devices Draft Guidance for Industry And Food And Drug Administration Staff. Retrieved April 26, 2016 from
⦁ FDA. (2016, April 20). Unique Device Identification - UDI. Retrieved April 26, 2016, from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/default.htm
⦁ ISO. (2011, October 15). ISO/IEC TR 29158:2011. Retrieved April 29, 2016 from http://www.iso.org/iso/catalogue_detail.htm?csnumber=45237
⦁ [Untitled image of example of a unique device identifier (UDI) on a medical label]. (2013, September 18). Retrieved April 29, 2016 from http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/UniqueDeviceIdentification/UDIBasics/default.htm
The content & opinions in this article are the author’s and do not necessarily represent the views of ManufacturingTomorrow
Comments (0)
This post does not have any comments. Be the first to leave a comment below.
Featured Product

Quickbase: The first application platform built for dynamic work
By connecting everything through a single source of truth, the Quickbase platform helps businesses mitigate risk, reduce waste, and cut down on unexpected costs. With automated workflows and granular permissions, the right people will have access to the right information.
